
The developed illustrative biophysical model and the corresponding software make it possible to directly analyze the reactivity of amyloid peptides and molecules that bind to them.

Our method include:
-Study of hereditary forms of Alzheimer's disease,
- Study of the stability and kinetics of aggregation,
- Increased stability of amyloid peptides,
-determination of key amino acid residues,
-selection of antibodies to amyloids
-Study of hereditary forms of Alzheimer's disease,
- Study of the stability and kinetics of aggregation,
- Increased stability of amyloid peptides,
-determination of key amino acid residues,
-selection of antibodies to amyloids
Study of hereditary cases of disease resistance reveals possible Alzheimer's treatment.
Treatment costs and number of cases
Currently, there are ~46.8 million dementia cases worldwide, with that number projected to reach 74.7 million in 2030 and over 131.5 million by 2050. Alzheimer's disease (AD) is a major public health problem in the world. Health care costs of dementia in 2015 surpassed $818 billion (USD), and this figure is estimated to be as high as $2 trillion by 2030.[1]
An example of the result of the action of various inherited mutations in genes, their quantity.
For example, Family members who carry a rare gene mutation called Presenilin 1 (PSEN1) E280A, have a 99.9% risk of developing early-onset Alzheimer's disease.
The researchers confirmed that the woman in this case carried the PSEN1 E280A mutation, which caused early-onset Alzheimer's in her other family members. However, she also had two copies of the APOE3ch gene variant, while no other affected family member carried two copies of this variant.
However, she did not develop signs of the disease until her seventies, nearly three decades after her expected age of onset. The researchers suspect that she may have been protected because in addition to the gene mutation causing early-onset Alzheimer's in her family, she also had two copies of the APOE3 Christchurch (APOE3ch) gene variant.
Imaging tests showed that the woman had only minor neurodegeneration. She did have large amounts of amyloid protein deposits, a hallmark of Alzheimer's disease, in her brain. But the amount of tau tangles, another hallmark of the disease, and the one more correlated with how thinking and memory are affected, was relatively low.
The researchers confirmed that the woman in this case carried the PSEN1 E280A mutation, which caused early-onset Alzheimer's in her other family members. However, she also had two copies of the APOE3ch gene variant, while no other affected family member carried two copies of this variant.
However, she did not develop signs of the disease until her seventies, nearly three decades after her expected age of onset. The researchers suspect that she may have been protected because in addition to the gene mutation causing early-onset Alzheimer's in her family, she also had two copies of the APOE3 Christchurch (APOE3ch) gene variant.
Imaging tests showed that the woman had only minor neurodegeneration. She did have large amounts of amyloid protein deposits, a hallmark of Alzheimer's disease, in her brain. But the amount of tau tangles, another hallmark of the disease, and the one more correlated with how thinking and memory are affected, was relatively low.
What type of Alzheimer's is hereditary?
Early-onset and late-onset familial Alzheimer disease is inherited in an autosomal dominant pattern, which means one copy of an altered gene in each cell is sufficient to cause the disorder. In most cases, an affected person inherits the altered gene from one affected parent. If a parent has a mutated gene that causes FAD, each child has a 50% chance of inheriting it. The presence of the gene means that the person will eventually develop Alzheimer's disease, usually in their 40s or 50s
Alzheimer's Disease Genetics
When a genetic variant increases disease risk but does not directly cause a disease, it is called a genetic risk factor. Identifying genetic variants may help researchers find the most effective ways to treat or prevent diseases such as Alzheimer's in an individual. This approach, called precision medicine, takes into account individual variability in genes, environment, and lifestyle for each person.
Genes and Alzheimer's Disease
The faulty gene can only be passed down directly from a parent who has familial Alzheimer's, it does not skip generations.
So far three genes have been linked to young onset inherited Alzheimer's disease. These genes are called:
- amyloid precursor protein (APP)
- presenilin 1 (PSEN1)
- presenilin 2 (PSEN2).
These genes are involved in the production of a protein called amyloid. If the gene is faulty, there can be an abnormal build-up of amyloid in the brain that causes clumps or 'plaques', a characteristic feature of Alzheimer's disease.
When a genetic variant increases disease risk but does not directly cause a disease, it is called a genetic risk factor. Identifying genetic variants may help researchers find the most effective ways to treat or prevent diseases such as Alzheimer's in an individual. This approach, called precision medicine, takes into account individual variability in genes, environment, and lifestyle for each person.
Genes and Alzheimer's Disease
The faulty gene can only be passed down directly from a parent who has familial Alzheimer's, it does not skip generations.
So far three genes have been linked to young onset inherited Alzheimer's disease. These genes are called:
- amyloid precursor protein (APP)
- presenilin 1 (PSEN1)
- presenilin 2 (PSEN2).
These genes are involved in the production of a protein called amyloid. If the gene is faulty, there can be an abnormal build-up of amyloid in the brain that causes clumps or 'plaques', a characteristic feature of Alzheimer's disease.
The figure1 shows a diagram of an amyloid peptide indicating hereditary mutations that have different distinctive features at the physiological level, and also have different characteristics in the studied physical interactions with other molecules (with amyloid peptides, oligomers, molecular complexes).
The figure1 shows those hereditary mutations that will be studied in detail for resistance, toxicity, aggregation kinetics, manifestations of Alzheimer's disease and other diseases.
The figure1 shows those hereditary mutations that will be studied in detail for resistance, toxicity, aggregation kinetics, manifestations of Alzheimer's disease and other diseases.

Fig.1. Amino acid sequence of the ABeta(11–42) peptide, with its missense mutations being indicated. AAR numbering is given in two variants: for the peptide itself and relative to its predecessor protein APP.
Scheme of the APP region, indicating known mutations
A.1.
MUTATIONS IN APP
APP encodes amyloid precursor protein, a transmembrane protein which is cleaved to form amyloidogenic Aβ peptides. Mutations in APP are associated with familial forms of early onset Alzheimer's disease as well as with Cerebral Amyloid Angiopathy (CAA). Pathogenic mutations generally alter processing by secretases, leading in an overall increase in Aβ production and/or a change in the ratio of specific Aβ peptides.
K16N
The mutation was described in an individual affected by a form of early onset dementia consistent with Alzheimer's disease. The patient presented with progressive deficits across several cognitive domains, including short-term memory, mathematical ability, and verbal fluency. (Kaden et al., 2012).
In vitro, this mutation makes APP a poor substrate for cleavage by α-secretase, with reduced production of total sAPP, and especially sAPPα. Although mutations in the Aβ region typically enhance Aβ toxicity, the Aβ42 K16N peptide was less harmful to neuroblastoma cells than wild-type Aβ42. However, the mutant peptide was highly toxic when mixed in equimolar amounts with wild-type Aβ, the expected situation in a patient heterozygous for the mutation. Similarly, although the mutant Aβ peptide formed predominantly low-n oligomers in vitro, when mixed with wild-type Aβ it aggregated into high-n oliogmers. The Aβ K16N peptide was also much less efficiently degraded by neprilysin (Kaden et al., 2012).
In vitro, this mutation makes APP a poor substrate for cleavage by α-secretase, with reduced production of total sAPP, and especially sAPPα. Although mutations in the Aβ region typically enhance Aβ toxicity, the Aβ42 K16N peptide was less harmful to neuroblastoma cells than wild-type Aβ42. However, the mutant peptide was highly toxic when mixed in equimolar amounts with wild-type Aβ, the expected situation in a patient heterozygous for the mutation. Similarly, although the mutant Aβ peptide formed predominantly low-n oligomers in vitro, when mixed with wild-type Aβ it aggregated into high-n oliogmers. The Aβ K16N peptide was also much less efficiently degraded by neprilysin (Kaden et al., 2012).
A21G APP A692G (Flemish)
Affected individuals were diagnosed with Alzheimer's disease or cerebral hemorrhage associated with cerebral amyloid angiopathy. The mean age at onset in this family was 45.7 ± 7.3 years (Hendriks et al., 1992).
Neuropathologic examination confirmed AD, congophilic angiopathy, and hemorrhagic infarction. In this family, dementia onset ranged from 39 to 54 years of age (Brooks et al., 2004).
Biological Effect
Aβ peptides carrying the Flemish mutation have relatively low aggregation propensity in vitro, including dimerization (Murakami et al., 2002; Meinhardt et al., 2007; Huet et al., 2006). Conformational changes are thought to explain the Flemish mutation's inhibitory effect on self-assembly and nucleation. However, fibril formation was promoted through interaction with gangliosides in the vascular wall, which may partly explain the propensity of Flemish Aβ to deposit in the vasculature (Yamamoto et al., 2005; Yagi-Utsumi and Dobson 2015). Flemish mutation A21G (A692G in full-length APP) leads to AD accompanied by extensive Aβ deposition
Neuropathologic examination confirmed AD, congophilic angiopathy, and hemorrhagic infarction. In this family, dementia onset ranged from 39 to 54 years of age (Brooks et al., 2004).
Biological Effect
Aβ peptides carrying the Flemish mutation have relatively low aggregation propensity in vitro, including dimerization (Murakami et al., 2002; Meinhardt et al., 2007; Huet et al., 2006). Conformational changes are thought to explain the Flemish mutation's inhibitory effect on self-assembly and nucleation. However, fibril formation was promoted through interaction with gangliosides in the vascular wall, which may partly explain the propensity of Flemish Aβ to deposit in the vasculature (Yamamoto et al., 2005; Yagi-Utsumi and Dobson 2015). Flemish mutation A21G (A692G in full-length APP) leads to AD accompanied by extensive Aβ deposition
E22G APP (Arctic mutation)
Aβ deposition in the brains was wide-spread and profuse. Virtually all parenchymal deposits were composed of non-fibrillar. Most cerebral cortical plaques appeared targetoid with centres containing C-terminally (beyond aa 40) and variably N-terminally truncated Aβ surrounded by coronas immunopositive for Aβx-42.
In Arctic AD brain differentially truncated abundant Aβ is deposited in plaques of variable numbers and shapes in different regions of the brain. The extracellular non-fibrillar Aβ does not seem to cause overt damage to adjacent neurons or to induce formation of neurofibrillary tangles, supporting the view that intracellular Aβ oligomers are more neurotoxic than extracellular Aβ deposits. Finally, similarly as the cotton wool plaques in AD resulting from exon 9 deletion in the presenilin-1 gene, the Arctic plaques induced only modest glial and inflammatory tissue reaction [5].
Neuropathology No signs of strokes or vascular lesions were found by brain imaging (Nilsberth et al., 2001). Neuropathology was available for only one mutation carrier and showed neuritic plaques and neurofibrillary tangles consistent with a diagnosis of AD (Kamino et al., 1992). Cortical PiB retention was very low in two Arctic mutation carriers compared with both noncarrier siblings and two individuals with other pathogenic mutations (APP Swedish and PSEN1 H163Y).
Arctic Aβ40 forms protofibrils at an increased propensity and faster rate compared with wild-type Aβ40 (Nilsberth et al., 2001).
In Arctic AD brain differentially truncated abundant Aβ is deposited in plaques of variable numbers and shapes in different regions of the brain. The extracellular non-fibrillar Aβ does not seem to cause overt damage to adjacent neurons or to induce formation of neurofibrillary tangles, supporting the view that intracellular Aβ oligomers are more neurotoxic than extracellular Aβ deposits. Finally, similarly as the cotton wool plaques in AD resulting from exon 9 deletion in the presenilin-1 gene, the Arctic plaques induced only modest glial and inflammatory tissue reaction [5].
Neuropathology No signs of strokes or vascular lesions were found by brain imaging (Nilsberth et al., 2001). Neuropathology was available for only one mutation carrier and showed neuritic plaques and neurofibrillary tangles consistent with a diagnosis of AD (Kamino et al., 1992). Cortical PiB retention was very low in two Arctic mutation carriers compared with both noncarrier siblings and two individuals with other pathogenic mutations (APP Swedish and PSEN1 H163Y).
Arctic Aβ40 forms protofibrils at an increased propensity and faster rate compared with wild-type Aβ40 (Nilsberth et al., 2001).
E22K APP E693K (Italian)
Neuropathology Neuroimaging showed small to large hematomas, subarachnoid bleeding, scars with hemosiderin deposits, small infarcts, and leukoaraiosis. Aβ immunoreactivity was detected in the walls of leptomeningeal and parenchymal vessels and in the neuropil. This disease is distinguished from Alzheimer's disease with CAA by the absence of neurofibrillary changes and neuritic plaques (Tagliavini et al., 1999; Bugiani et al., 2010).
CAA caused by the Italian or Dutch Aβ variants is uniquely characterized by deposition of Aβ in the smooth muscle cells surrounding the cerebral vasculature as compared to the typical Aβ pathology in sporadic AD, which is found mostly in brain parenchyma
The absence of neuritic plaques and tangles made it possible to distinguish the disease from familial AD with CAA and to call it hereditary cerebral hemorrhage with amyloidosis (HCHWA).
Biological Effect It is possible that this variant affects the conformation of Aβ peptides changing their rate of fibrillization. Indeed, a variant in this same position and associated with a similar phenotype, E693Q, accelerates polymerization into protofibrils and fibrils.
CAA caused by the Italian or Dutch Aβ variants is uniquely characterized by deposition of Aβ in the smooth muscle cells surrounding the cerebral vasculature as compared to the typical Aβ pathology in sporadic AD, which is found mostly in brain parenchyma
The absence of neuritic plaques and tangles made it possible to distinguish the disease from familial AD with CAA and to call it hereditary cerebral hemorrhage with amyloidosis (HCHWA).
Biological Effect It is possible that this variant affects the conformation of Aβ peptides changing their rate of fibrillization. Indeed, a variant in this same position and associated with a similar phenotype, E693Q, accelerates polymerization into protofibrils and fibrils.
E22Q APP E693Q (Dutch)
The discovery of this mutation was an early demonstration that a variant in the APP gene could cause severe amyloid deposition (Levy et al., 1990; van Broeckhoven et al., 1990; Fernandez-Madrid et al., 1991).
Neuropathology This mutation is associated with severe amyloid deposition in cerebral vessels, hemorrhages, and diffuse plaques in brain parenchyma (Timmers et al., 1990). Extensive Aβ accumulates in the cerebral vessels, especially the meningeal arteries and the cerebro-cortical arterioles. In people with HCHWA-D, the amount of CAA is correlated with dementia, whereas parenchymal plaque density and intraneuronal neurofibrillary tangles are not (Natté et al., 2001).
Biological Effect This mutation results in the accumulation of Aβ in cerebral vessel walls. This pathology leads to cell death and loss of vessel wall integrity, which in turn makes the vessels prone to obstruction and rupture, manifesting clinically as hemorrhages and infarcts. In vitro, this mutation accelerates Aβ aggregation, leading to increased fibril formation (Wisniewski et al., 1991).This mutation is associated with high levels of β-sheet conformation and induction of apoptosis in cerebral endothelial cells compared with wild-type Aβ (Miravalle et al., 2000)
Neuropathology This mutation is associated with severe amyloid deposition in cerebral vessels, hemorrhages, and diffuse plaques in brain parenchyma (Timmers et al., 1990). Extensive Aβ accumulates in the cerebral vessels, especially the meningeal arteries and the cerebro-cortical arterioles. In people with HCHWA-D, the amount of CAA is correlated with dementia, whereas parenchymal plaque density and intraneuronal neurofibrillary tangles are not (Natté et al., 2001).
Biological Effect This mutation results in the accumulation of Aβ in cerebral vessel walls. This pathology leads to cell death and loss of vessel wall integrity, which in turn makes the vessels prone to obstruction and rupture, manifesting clinically as hemorrhages and infarcts. In vitro, this mutation accelerates Aβ aggregation, leading to increased fibril formation (Wisniewski et al., 1991).This mutation is associated with high levels of β-sheet conformation and induction of apoptosis in cerebral endothelial cells compared with wild-type Aβ (Miravalle et al., 2000)
D23N APP IOWA
Affected individuals presented with a hereditary syndrome involving hemorrhagic stroke, dementia, leukoencephalopathy, and occipital calcifications. Age of onset in this family was 58 to 66 years.
Overall, symptom onset in this family ranged from 38 to 47 years of age.
Neuropathology Neuropathological examination of the original Iowa proband revealed severe cerebral amyloid angiopathy, widespread neurofibrillary tangles, and abundant Aβ40 in plaques. The proband and an affected brother also had microscopic hemorrhagic lesions and cortical calcifications in the occipital lobe (Grabowski et al., 2001). Neuropathological examination in the Irish patient, LM, confirmed a large occipital hemorrhage with severe amyloid angiopathy of meningeal, cerebro-cortical, and cerebellar parenchymal arteries and veins.
Overall, symptom onset in this family ranged from 38 to 47 years of age.
Neuropathology Neuropathological examination of the original Iowa proband revealed severe cerebral amyloid angiopathy, widespread neurofibrillary tangles, and abundant Aβ40 in plaques. The proband and an affected brother also had microscopic hemorrhagic lesions and cortical calcifications in the occipital lobe (Grabowski et al., 2001). Neuropathological examination in the Irish patient, LM, confirmed a large occipital hemorrhage with severe amyloid angiopathy of meningeal, cerebro-cortical, and cerebellar parenchymal arteries and veins.
L34V APP L705V
The proband and three affected cousins carried the mutation while an unaffected 74-year-old cousin did not, suggesting cosegregation of the mutation with disease.
At age 62, the carrier suffered multiple, large intracerebral hemorrhages over a four-month period, the last of which was fatal. Notably, except for brief periods immediately following each hemorrhage, her cognition remained intact. As in the original report of this mutation, there is a family history of hemorrhagic strokes in individuals in their 40s to 70s that is consistent with an autosomal dominant pattern of inheritance.
Neuropathology. The amyloid deposition appeared to selectively affect vessel walls. There was no evidence of Aβ parenchymal deposits (either diffuse or neuritic plaques), nor were neurofibrillary tangles or dystrophic neurites observed (Obici et al., 2005).
At age 62, the carrier suffered multiple, large intracerebral hemorrhages over a four-month period, the last of which was fatal. Notably, except for brief periods immediately following each hemorrhage, her cognition remained intact. As in the original report of this mutation, there is a family history of hemorrhagic strokes in individuals in their 40s to 70s that is consistent with an autosomal dominant pattern of inheritance.
Neuropathology. The amyloid deposition appeared to selectively affect vessel walls. There was no evidence of Aβ parenchymal deposits (either diffuse or neuritic plaques), nor were neurofibrillary tangles or dystrophic neurites observed (Obici et al., 2005).
APP G709S
This variant was detected in one out of 188 individuals with Parkinson's disease with dementia (Schulte et al., 2015). This individual presented with resting tremor, and also developed bradykinesia, rigor, postural instability, and dementia. An uncle also had PD. The variant was absent in 188 PD cases without dementia and 376 controls.
Mapping the Potential Energy of Electrostatic Interaction
Mapping the Potential Energy of Electrostatic Interaction between Amino Acid Residues of Interacting Peptides Forming Oligomers and High-Molecular Complexes
The potential energy map of the electrostatic interaction makes it possible to determine the energy contribution of each amino acid residue to the formation of the amyloid dimer.
The figure below shows a map in the case of the formation of an amyloid dimer, however, the potential energy can be calculated for any, arbitrarily complex biological complex.
The peaks in the scheme indicate amino acid residues that are characterized by a large value of the potential energy of interaction.
The figure below shows a map in the case of the formation of an amyloid dimer, however, the potential energy can be calculated for any, arbitrarily complex biological complex.
The peaks in the scheme indicate amino acid residues that are characterized by a large value of the potential energy of interaction.

Fig.2. Three-dimensional map of the potential energy of pairwise electrostatic interaction between two wild-type amyloid peptides (upper figure) and taking into account mutations (lower figure)
Next, we will consider the illustrative results obtained for the change in the stability of amyloid dimers taking into account various mutations in amyloid peptides.
The figures below show the numerical results obtained for various mutations and a brief description of their physical properties, which varied depending on changes in the resistance of amyloids at the level of dimeric complexes.
The figures below show the numerical results obtained for various mutations and a brief description of their physical properties, which varied depending on changes in the resistance of amyloids at the level of dimeric complexes.
Stabilization of amyloid peptides at the level of dimeric complexes
We have developed a method for increasing the stability of amyloid dimers in order to reduce their ability to enter into further biochemical reactions with the formation of toxic oligomers.
Numerical calculations are presented in the form of a graphical description, which clearly allows you to see an increase or decrease in the stability of dimeric amyloid complexes. Our proposed method allows us to vary the range of stability of dimeric complexes by substitutions of key amino acid residues.
Numerical calculations are presented in the form of a graphical description, which clearly allows you to see an increase or decrease in the stability of dimeric amyloid complexes. Our proposed method allows us to vary the range of stability of dimeric complexes by substitutions of key amino acid residues.

Fig.3.Values for stability indicator lg(cond(W)) a) that were obtained by considering the interaction of wild form wtAB and interaction of mutant peptide forms mutAB from Table 1, calculated lg(cond(W)) value for random mutations (b), calculated ∆H value for inherited mutations (c), comparative analysis of lg(cond(W)) value for inherited mutations including D23N replacement (d).
The stability of biological complex is plotted in Fig. 3. The mutation names and the way the structures of a higher order are formed are indicated in the plots. A large arrow between the plots indicates the direction for values characteristic of the formation of structures with a higher molecular weight such as oligomers, protofibrils, and fibrils.
A thick line at a level of 5.53 arbitrarily separates structures that tend to (the region below the line).
On the strength of experimental data on missense mutations and their biological effect, we draw a separation line at a level of 5.53 that separates mutations in peptides leading to enhanced formation of higher order structures from mutations in ABeta peptides exhibiting a reduced capacity to form high-molecular weight structures.
Two vertical arrows pointing away from the line drawn at 5.53 indicate directions in the regions characterized by the lower (arrow up) or higher (arrow down) stability of dimer complexes. The present grading was obtained and tested for a three-dimensional complex from the PDB:2MXU database [12].
A thick line at a level of 5.53 arbitrarily separates structures that tend to (the region below the line).
On the strength of experimental data on missense mutations and their biological effect, we draw a separation line at a level of 5.53 that separates mutations in peptides leading to enhanced formation of higher order structures from mutations in ABeta peptides exhibiting a reduced capacity to form high-molecular weight structures.
Two vertical arrows pointing away from the line drawn at 5.53 indicate directions in the regions characterized by the lower (arrow up) or higher (arrow down) stability of dimer complexes. The present grading was obtained and tested for a three-dimensional complex from the PDB:2MXU database [12].
If the dimeric complex is stable, then the formation of high molecular weight structures was much slower.
This was due to the fact that the stable amyloid peptides were in no hurry to enter into chemical reactions with other amyloid peptides to achieve equilibrium.
This was due to the fact that the stable amyloid peptides were in no hurry to enter into chemical reactions with other amyloid peptides to achieve equilibrium.

K16N-K16N
K16N-WT
the Aβ42 K16N peptide was less harmful to neuroblastoma cells than wild-type Aβ42
the mutant Aβ peptide formed predominantly low-n oligomers in vitro
the mutant Aβ peptide formed predominantly low-n oligomers in vitro
the mutant peptide was highly toxic when mixed in equimolar amounts with wild-type Aβ mixed with wild-type Aβ it
aggregated into high-n oliogmers.
A21G-A21G
(Flemish)
(Flemish)
a) Aβ peptides carrying the Flemish mutation have relatively low aggregation propensity in vitro, including dimerization
b) Conformational changes are thought to explain the Flemish mutation's inhibitory effect on self-assembly and nucleation.
b) Conformational changes are thought to explain the Flemish mutation's inhibitory effect on self-assembly and nucleation.
E22K- E22K
(Italian)
(Italian)
a) Arctic Aβ40 forms protofibrils at an increased propensity and faster rate compared with wild-type Aβ40
b) In Arctic AD brain differentially truncated abundant Aβ is deposited in plaques of variable numbers and shapes in different regions of the brain.
b) In Arctic AD brain differentially truncated abundant Aβ is deposited in plaques of variable numbers and shapes in different regions of the brain.
CAA caused by the Italian or Dutch Aβ variants is uniquely characterized by deposition of Aβ in the smooth muscle cells surrounding the cerebral vasculature
E22Q- E22Q
(Dutch)
(Dutch)
E22Q-E22Q
(Dutch)
(Dutch)
a) The discovery of this mutation was an early demonstration that a variant in the APP gene could cause severe amyloid deposition.
b) Extensive Aβ accumulates in the cerebral vessels, especially the meningeal arteries and the cerebro-cortical arterioles.
c) In vitro, this mutation accelerates Aβ aggregation, leading to increased fibril formation.
d) This mutation is associated with high levels of β-sheet conformation and induction of apoptosis in cerebral endothelial cells compared with wild-type Aβ
b) Extensive Aβ accumulates in the cerebral vessels, especially the meningeal arteries and the cerebro-cortical arterioles.
c) In vitro, this mutation accelerates Aβ aggregation, leading to increased fibril formation.
d) This mutation is associated with high levels of β-sheet conformation and induction of apoptosis in cerebral endothelial cells compared with wild-type Aβ
[K16N]Aβ42 was found to be less toxic than WT Aβ42 at equal concentrations. However, a mixture of the two adding up to the same concentration was equally or more toxic than WT Aβ42 alone
Fig.5. Oligomerization and toxicity of K16N substituted Aβ peptides.
(1) SH-SY5Y cells were incubated for 12 hours with either 2 μM freshly dissolved peptides (load) of Aβ40 (A) or Aβ42 (B), or oligomers (2–20x) obtained by SEC.
(2) Primary hippocampal neurons were incubated for 48 hours with 2 μM freshly dissolved peptides. Toxicity was determined by percentage of living cells compared to untreated control cells (n = 4–8). The data are presented as mean ± SEM. (*p < 0.001, **p < 0.0001). Reprinted with permission from Kaden et al [97].
(1) SH-SY5Y cells were incubated for 12 hours with either 2 μM freshly dissolved peptides (load) of Aβ40 (A) or Aβ42 (B), or oligomers (2–20x) obtained by SEC.
(2) Primary hippocampal neurons were incubated for 48 hours with 2 μM freshly dissolved peptides. Toxicity was determined by percentage of living cells compared to untreated control cells (n = 4–8). The data are presented as mean ± SEM. (*p < 0.001, **p < 0.0001). Reprinted with permission from Kaden et al [97].
[more stable]
It is characterized by high thermodynamic stability compared to other dimers, as well as the least tendency to enter into biochemical reactions to achieve equilibrium. As one of the biological consequences of finding a dimer with high thermodynamic stability, there is a low reactivity, a reduced rate of forming high-molecular complexes, including fibrils and oligomers.
[less stable]
It is characterized by reduced thermodynamic stability compared to other dimers, as well as the greatest tendency to enter into biochemical reactions to achieve equilibrium. As one of the biological consequences of the definition of a dimer with low thermodynamic stability is a high reactivity, an increased rate of formation of high-molecular complexes, including fibrils and oligomers.
Analysis of the stability of amyloid peptides taking into account hereditary mutations. The calculation was performed using the software developed by our team
K16N+K16N _____5,5264
wt-wt ____________5,5372
K16N+wt ________5,5312
wt-wt ____________5,5372
K16N+wt ________5,5312
E22G- E22G
(Arctic)
(Arctic)
[ABeta]2 ______lg(cond(w))
[more stable]
[less stable]
[middle value]
E22K-E22K__ _____5.55431
WT-WT ___________5,5372
WT-WT ___________5,5372
[ABeta]2 ______lg(cond(w))
[more stable]
[less stable]
E22G-E22G_ _____5.5492
wt-wt ____________5,5372
wt-wt ____________5,5372
[ABeta]2 ________lg(cond(w))
[more stable]
[less stable]
E21G-E21G_ _____5.51966
wt-wt ____________5,5372
wt-wt ____________5,5372
[ABeta]2 ________lg(cond(w))
[more stable]
[less stable]
E22K-E22K__ _____5.54094
WT-WT ___________5,5372
WT-WT ___________5,5372
[ABeta]2 ______lg(cond(w))
[more stable]
[less stable]
E22K-E22K__ _____5.55431
WT-WT ___________5,5372
WT-WT ___________5,5372
[ABeta]2 ______lg(cond(w))
[more stable]
[less stable]
E22K-E22K__ _____5.55431
WT-WT ___________5,5372
WT-WT ___________5,5372
[ABeta]2 ______lg(cond(w))
[more stable]
[less stable]


(1)
(2)
K16N+K16N ____________5,5264
wt-wt ___________________5,5372
K16N+wt _______________5,5312
E21G-E21G _____________5,51966
wt-wt ___________________5,5372
E22K-E22K _____________ 5,55431
WT-WT _________________ 5,5372
E22K-E22K _____________5,54094
WT-WT _________________5,5372
wt-wt ___________________5,5372
K16N+wt _______________5,5312
E21G-E21G _____________5,51966
wt-wt ___________________5,5372
E22K-E22K _____________ 5,55431
WT-WT _________________ 5,5372
E22K-E22K _____________5,54094
WT-WT _________________5,5372
Direct Correlation Results Between Numerical Values and Biological Effects
B.1.
In addition to investigating genetic mutations, we also performed a calculation for random mutations in amyloid peptides and analysed their effect on the half-life of aggregation. Estimated data of lg(cond(W)) value for such mutations are shown in Fig.6 b. The structure with the D23N substitution in the two amyloid peptides represents an antiparallel interaction in the formation of dimer, as opposed to the wild-type structure, whose formation is due to the parallel coupling of amyloid peptides.

Fig.7 Two amyloid peptide structures, with the upper structure corresponding to an interaction between wild-type amyloid peptides, and the lower structure corresponding to an interaction between two mutant forms of D23N peptides, with them forming a dimer by combining into antiparallel structures.
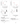
Figure 6: Correlation scheme for physical quantities such as lg(cond(W)) and ∆H. The value of lg(cond(W)) characterizes the stability of the biological complex, and ∆H characterizes the orderliness of the system as the system transitions from one state to another, in this case from the state of the interaction of wild-type amyloid dimers into a system of dimers given the substitutions of the selected amino acid residues.
We will pay special attention to the modified amyloid peptide pair mAβ(11−42)2 (D23N). In this dimer, two symmetric substitutions of D23N were performed. The numerical results are shown in Fig.d extreme value. As can be seen from the presented graphs, the replacement of D23N in the two polypeptide chains results in a sharp decrease in lg(cond(W)), while the ∆H values are conversely in the positive region of values (see Fig.6c). At the same time, the extreme values in the diagram d change significantly (contrast) with the rest of the values in the graph a. The necessary minimum requirement to indicate a significant disruption of molecular bounds and the transition of the system to a new position is the absence of co-directional change in the two quantities lg(cond(W)) and ∆H, which we observe in Fig.6 c and Fig. 6d. The structures of the two wild-type dimers when replacing D23N are shown in Fig.7 .
Method for determining conformational rearrangement by computational methods.
C.1.
Determination of modified amyloid peptides characterized by the greatest thermodynamic stability.
The second part will be a logical continuation of the search for stable dimeric complexes characterized by thermodynamic stability and a longer aggregation time.

Fig.8. Comparison of experimental and calculated values that characterize the stability of amyloid peptides and their ability to enter into biochemical processes (self-aggregation), depending on the substitutions performed.
Upper graph a) was obtained from the results of the experimental data ( Kinetic aggregation data was kindly provided by Sara Snogerup Linse, Department of Biochemistry and Structural Biology, Center for Molecular Protein Science, CMPS, LUND UNIVERSITY) at a concentration of 1.1 μM. The time of half completion, t1/2, for the serine mutants in comparison with the WT Aβ42 in 20 mM sodium phosphate and 200 μM EDTA at pH 8.0 is shown in the upper graph.
Below them b), c),d) are the corresponding calculated stability data for dimeric complexes taking into account the performed substitutions. At the same time, for clarity of viewing and comparison, the reciprocal value 1/lg(cond(W)) is given.
Thus, we see a relationship between the stability of dimeric modified amyloid peptides and the half-life of their self-aggregation.
Upper graph a) was obtained from the results of the experimental data ( Kinetic aggregation data was kindly provided by Sara Snogerup Linse, Department of Biochemistry and Structural Biology, Center for Molecular Protein Science, CMPS, LUND UNIVERSITY) at a concentration of 1.1 μM. The time of half completion, t1/2, for the serine mutants in comparison with the WT Aβ42 in 20 mM sodium phosphate and 200 μM EDTA at pH 8.0 is shown in the upper graph.
Below them b), c),d) are the corresponding calculated stability data for dimeric complexes taking into account the performed substitutions. At the same time, for clarity of viewing and comparison, the reciprocal value 1/lg(cond(W)) is given.
Thus, we see a relationship between the stability of dimeric modified amyloid peptides and the half-life of their self-aggregation.
We explain it this way: the more stable the dimeric complex is, the slower it will enter into new biochemical processes.
The next phase of research is in progress.
Next stages of research
The study of hereditary forms of mutations leading to early and late forms of dementia, as well as providing protective properties against the formation of plaques.
- Hight Quality amyloid peptidesDevelopment of amyloid peptides that are characterized by high thermodynamic stability, demonstrating increased time of aggregation kinetics (Done. Contact the authors for information)
- Hereditary forms of ADStudy of various schemes of biochemical reactions involving various hereditary forms of amyloid peptides. The construction of biochemical schemes is carried out in three stages, see the section "Biomarkers".
- Individual Approach and Personalized medicineIdentification of differences in the passage of biochemical reactions involving hereditary forms of amyloid peptides.
- Changing biochemical pathways.Introduction and analysis into biochemical pathways of peptides that meet the requirements of high kinetic and thermodynamic stability.
- Antibodies to amyloid peptidesSelection of flexible chains of antibodies both to amyloid peptides and to intermediate targets determined in biochemical chains. For selection of antibody flexible chains, see "Antibody Development".

Additional Information
Additional calculations for the search for peptides with the highest stability index,
i.e. minimum value lg(cond(W)).
[Go to full screen PC mode]
i.e. minimum value lg(cond(W)).
[Go to full screen PC mode]

Ala21
Ala30
Ala42
ABeta(11-42)
ABeta(11-42)


Ala21
Ala30
Ala42
ABeta(11-42)
ABeta(11-42)
Ala42
Ala30
Ala21



ABeta(11-42)
Ala42
Ala30
Ala21
ABeta(11-42)
PDB:5KK3
[mutABeta(11-42)]-[mutABeta (11-42)]
[mutABeta(11-42)]-[mutABeta (11-42)]
PDB:2MXU
[mutABeta(11-42)]-[mutABeta (11-42)]
[mutABeta(11-42)]-[mutABeta (11-42)]
PDB:2MXU
[wtABeta(11-42)]-[mutABeta (11-42)]
[wtABeta(11-42)]-[mutABeta (11-42)]
5,462
5,40


PDB:5KK3
[mutABeta(11-42)]-[mutABeta (11-42)]
[mutABeta(11-42)]-[mutABeta (11-42)]
5,39
5,605
PDB:2MXU
[mutABeta(11-42)]-[mutABeta (11-42)]
[mutABeta(11-42)]-[mutABeta (11-42)]




5,637
PDB:2MXU
[wtABeta(11-42)]-[mutABeta (11-42)]
[wtABeta(11-42)]-[mutABeta (11-42)]
PDB:5KK3
[wtABeta(11-42)]-[mutABeta (11-42)]
[wtABeta(11-42)]-[mutABeta (11-42)]
[wtABeta(11-42)]
[mutABeta(11-42)]
[wtABeta(11-42)]
[mutABeta(11-42)]
21ALA
30ALA
21ALA
30ALA

PDB:5KK3
[wtABeta(11-42)]-[mutABeta (11-42)]
[wtABeta(11-42)]-[mutABeta (11-42)]
[wtABeta(11-42)]
[mutABeta(11-42)]
21ALA
30ALA
5,483




PDB:2MXU
[wtABeta(11-42)]-[mutABeta (11-42)]
[wtABeta(11-42)]-[mutABeta (11-42)]
PDB:5kk3
[wtABeta(11-42)]-[mutABeta (11-42)]
[wtABeta(11-42)]-[mutABeta (11-42)]
5,495
5,647
[mutABeta(11-42)]
[wtABeta(11-42)]
21ALA
42ALA
[mutABeta(11-42)]
[wtABeta(11-42)]
21ALA
42ALA



PDB:2MXU
[wtABeta(11-42)]-[mutABeta (11-42)]
[wtABeta(11-42)]-[mutABeta (11-42)]
PDB:2MXU
[mutABeta(11-42)]-[mutABeta (11-42)]
[mutABeta(11-42)]-[mutABeta (11-42)]
[wtABeta(11-42)]
[mutABeta(11-42)]
[mutABeta(11-42)]
[mutABeta(11-42)]
21ALA
42ALA
30ALA
28LYS
42ALA
42ALA
30ALA
30ALA
28LYS
21ALA
21ALA
This graph illustrates the modified amyloid peptides found in this work, which form the most stable dimers in comparison with the above wild-type amyloid peptides and modified peptides.
PDB:2MXU
[mutABeta(11-42)]2
[mutABeta(11-42)]2
PDB:2MXU
[wtABeta(11-42)]-[mutABeta (11-42)]
[wtABeta(11-42)]-[mutABeta (11-42)]
21ASN_28ASN_30HIS_42HIS ______5,35363
21ASN_28MET_30HIS_42MET _____5,35529
21ASN_28ASP_30ASN_42ASP ______5,4602
21HIS_28SER_30ASN_42HIS ______ 5,35847
21MET_28LEU_30ASN_42HIS _____ 5,36767
21MET_28GLN_30ASN_42ASP _____5,36414
21MET_28MET_30PHE_42PHE ____ 5,38547
21ASN_28MET_30HIS_42MET _____5,35529
21ASN_28ASP_30ASN_42ASP ______5,4602
21HIS_28SER_30ASN_42HIS ______ 5,35847
21MET_28LEU_30ASN_42HIS _____ 5,36767
21MET_28GLN_30ASN_42ASP _____5,36414
21MET_28MET_30PHE_42PHE ____ 5,38547

21ASN_28ASN_30HIS_42HIS
21ASN_28ASN_30HIS_42HIS
The area of positive values of potential electrostatic energy

21ASN_28ASN_30HIS_42HIS
21ASN_28ASN_30HIS_42HIS
The area of negative values of potential electrostatic energy
PDB:2MXU
[mutABeta(11-42)]2
[mutABeta(11-42)]2
lg(cond(W))
