
Intermediate results
detection of BRAF mutations that may cause resistance to targeted therapy
Nevi and Melanomas
Genetic susceptibility to nevi may affect the risk of developing melanoma, since common and atypical nevi are the main host risk factors implicated in the development of cutaneous melanoma.
What is the treatment for BRAF mutation?
BRAF inhibitors are drugs that can shrink or slow the growth of metastatic melanoma in people whose tumors have a BRAF mutation. BRAF inhibitors include vemurafenib (Zelboraf®), dabrafenib (Tafinlar®), and encorafenib (Braftovi®).
What is the survival rate of BRAF V600E?
Median progression free survival (mPFS) and objective response rate (ORR) of patients with BRAFV600E mutation was 7.3 months (95%CI: 4.6–9.1 months) and 30.1%, while patients with non-V600E mutations had an mPFS of 7.6 months (95%CI: 6.4–12.5 months) and an ORR of 37.5%
What is the mechanism of resistance to Vemurafenib?
Mechanisms of resistance to vemurafenib. BRAF(V600E) mutant tumors have constitutive activation of MEK and ERK (MAPK) signaling, which is inhibited by vemurafenib. Activating mutations (*) in NRAS or KRAS (1) or CRAF amplification (2) lead to activation of CRAF/MEK/ERK and resistance to RAF inhibition.
BRAF inhibitor resistance depends on oncogenic signaling through reactivation of MAPK/Erk or activation of PI3K/Akt, which may be acquired by directly affecting genes in each pathway, by upregulation of receptor tyrosine kinases, or by affecting downstream signaling.
Price list for Line Product
Main categories of molecular complexes with small inhibitors or atoms, and a list of prices for calculating each molecular complex.
Features
Set one
Set two
Set Three
Set Four
[Monomer+Inhibitor] or Atom
up to 10 mutations
up to 40 mutations
up to 150 mutations
up to 250 mutations
[3-4weeks]
[3-4weeks]
up to TWO inhibitors
[Dimer+Inhib.1+Inhib.2] or Atoms
up to 30 mutations
up to 100 mutations
up to 150 mutations
[3-5 weeks]
[3-5 weeks]
up to 4 inhibitors
[Tetramer+Several Inh-s]
[Trimer+Several Inhib-s]
[Trimer+Several Inhib-s]
up to 10 mutations
up to 50 mutations
[6-8 weeks]
[6-8 weeks]
[pentamer+Several Inh-s]
[any protein complex +Inh.]
[any protein complex +Inh.]
up to 10 mutations
Numerous cancers depend on this pathway for proliferation, as demonstrated by a diverse set of oncogenic alterations including mutations, increased gene copy numbers or enhanced expression of pathway components. The majority of variations occur at the level of Ras or RAF, and the BRAF isoform is the most frequently mutated protein kinase, with the V600E variation (BRAFV600E) being the most prevalent [Negative regulation of RAF kinase activity by ATP is overcome by 14-3-3-induced dimerization]
Melanocytes are melanin-producing cells that typically reside in the basal layer of the epidermis. When the MAPK pathway is activated, for example by a UV-induced mutation in the BRAF (mainly V600E) or NRAS genes, melanocytes undergo proliferation leading to the development of a nevus, then entering a state of senescence and stabilizing the structure of the nevus for many years.

The major downstream targets of ERK1/2 in the MAPK pathway. ERK regulates both cytosolic targets and nuclear transcription factors, thus promoting proliferation, survival and other malignant phenotypes.[Targeting ERK, an Achilles' Heel of the MAPK pathway, in cancer therapy]

Activation and feedback regulation of the MAPK pathway. The classical MAPK pathway is activated in human tumors by upstream [Targeting ERK, an Achilles' Heel of the MAPK pathway, in cancer therapy]
The mitogen-activated protein kinases (MAPK) pathway, often known as the RAS-RAF-MEK-ERK signal cascade, functions to transmit upstream signals to its downstream effectors to regulate physiological process such as cell proliferation, differentiation, survival and death. As the most frequently mutated signaling pathway in human cancer, targeting the MAPK pathway has long been considered a promising strategy for cancer therapy. Substantial efforts in the past decades have led to the clinical success of BRAF and MEK inhibitors.
Table 1
The role of the main melanoma-susceptibility genes in melanoma risk.
The role of the main melanoma-susceptibility genes in melanoma risk.
Genes
____________________________Prevalence______ Penetrance_______ Risk of Melanoma __________Reference
____________________________Prevalence______ Penetrance_______ Risk of Melanoma __________Reference
CDKN2A ______________Low_________________ High__________________ 35- to 70-fold
CDK4_________________Low______________High_____________________unknown
BAP1 __________________Low______________High_____________________unknown
POT1
ACD ______ ____________Low______________High_____________________unknown
TERF2IP
TERT
ACD ______ ____________Low______________High_____________________unknown
TERF2IP
TERT
MITF___________________Low_________Moderate to low_________3- to 5-fold
MC1R_______________Moderate____Moderate to low___________3-fold
DOCK8, KITLG, OCA2, MTAP, PLA2G6, SLC45A2, IRF4, OBFC1, FTO, PARP1
High______________Low______________________>3-fold *
* In the polygenic risk score model.
CDKN2A and its binding partner CDK4 were the first melanoma genes to be identified as melanoma-predisposing genes, though mutations in these loci have only been found in 20–45% of familial melanoma cases. CDKN2A (Cyclin-Dependent Kinase Inhibitor 2A) at 9p21 encodes two distinct proteins by alternative splicing (p16INK4A and p14ARF), both involved in cell-cycle regulation.
CDK4 (Cyclin-Dependent Kinase 4 in 12q14) is an oncogene involved in the same pathway of p16INK4A [37]. On the basis of available data, and similar to CDKN2A, mutations in CDK4 are also associated with a high number of atypical nevi [12,37], and penetrance is estimated at 74%
CDK4 (Cyclin-Dependent Kinase 4 in 12q14) is an oncogene involved in the same pathway of p16INK4A [37]. On the basis of available data, and similar to CDKN2A, mutations in CDK4 are also associated with a high number of atypical nevi [12,37], and penetrance is estimated at 74%
ERK1 (p44) and ERK2 (p42) are the proteins encoded by two splice variants of the same gene, and members of the MAPK superfamily also include ERK3/4, ERK5, ERK7/8, Jun N-terminal kinase (JNK)1/2/3 and p38,α,β and γ
(ERK6) and δ all of which have been shown to play roles in cancer.
Previous studies have suggested that ERK1 and ERK2 may have distinct functions for proliferation, and RAS-induced transformation requires ERK2 rather than ERK1.
ERK1 has been discovered to antagonistically compete with ERK2 for MEK, which results in a weakening of ERK2 signaling. Targeting ERK, an Achilles' Heel of the MAPK pathway, in cancer therapy. Available from: https://www.researchgate.net/publication/323220502_Targeting_ERK_an_Achilles%27_Heel_of_the_MAPK_pathway_in_cancer_therapy
(ERK6) and δ all of which have been shown to play roles in cancer.
Previous studies have suggested that ERK1 and ERK2 may have distinct functions for proliferation, and RAS-induced transformation requires ERK2 rather than ERK1.
ERK1 has been discovered to antagonistically compete with ERK2 for MEK, which results in a weakening of ERK2 signaling. Targeting ERK, an Achilles' Heel of the MAPK pathway, in cancer therapy. Available from: https://www.researchgate.net/publication/323220502_Targeting_ERK_an_Achilles%27_Heel_of_the_MAPK_pathway_in_cancer_therapy
MEK is a central component in the MAPK signaling cascade. MEK1 and MEK2 are tyrosine (Tyr) and serine (Ser)/ threonine
(Thr) dual-specificity kinases and share approximately 80% similarity. Within the MAPK pathway, MEK1/2 are phosphorylated and
activated by RAF
Targeting ERK, an Achilles' Heel of the MAPK pathway, in cancer therapy. Available from: https://www.researchgate.net/publication/323220502_Targeting_ERK_an_Achilles%27_Heel_of_the_MAPK_pathway_in_cancer_therapy
(Thr) dual-specificity kinases and share approximately 80% similarity. Within the MAPK pathway, MEK1/2 are phosphorylated and
activated by RAF
Targeting ERK, an Achilles' Heel of the MAPK pathway, in cancer therapy. Available from: https://www.researchgate.net/publication/323220502_Targeting_ERK_an_Achilles%27_Heel_of_the_MAPK_pathway_in_cancer_therapy
Activation and feedback regulation of the MAPK pathway. The classical MAPK pathway is activated in human tumors by upstream receptor tyrosine kinases (RTK) or by mutations in RAS, BRAF, and MEK1. RTKs activate RAS by recruiting adaptor proteins (e.g., GRB-2) and exchange factors (e.g., SOS). RAS activation promotes the formation of RAF dimers, which activate MEK-ERK cascade through phosphorylation. ERK pathway activity is regulated by negative feedback at multiple levels, including the transcriptional activation of DUSP proteins that negatively regulate the pathway. ERK also phosphorylates and thus regulates CRAF and MEK activity directly. ERK, or its immediate substrate
RSK, also phosphorylates SOS at several residues, inhibiting its activity and thus negatively regulating RAS activity
Targeting ERK, an Achilles' Heel of the MAPK pathway, in cancer therapy. Available from: https://www.researchgate.net/publication/323220502_Targeting_ERK_an_Achilles%27_Heel_of_the_MAPK_pathway_in_cancer_therapy
RSK, also phosphorylates SOS at several residues, inhibiting its activity and thus negatively regulating RAS activity
Targeting ERK, an Achilles' Heel of the MAPK pathway, in cancer therapy. Available from: https://www.researchgate.net/publication/323220502_Targeting_ERK_an_Achilles%27_Heel_of_the_MAPK_pathway_in_cancer_therapy
These results on the site are for guidance only and are not final as additional physical parameters need to be verified.
1. The fraction of non-dissociated molecules after the reaction and concentration protein-ligand complex
2. Entropy Change. The case of a one-dimensional normal distribution
3. Entropy Change. The case of multinormal distribution
4. Dissociation Constant: Kd, Mol/L
5. Enthalpy change: delta (P), J
6. The thermal dissociation
7. Potential energy of electrostatic interaction between all amino acid residues taken in pairs: Wp, J
8. Potential energy of the lower vibrational level: Wp1, J
9. Potential energy of the upper vibrational level: Wp2, J
10. Step-by-step verification of the PDB file structure (link: http://biomarker.co.il/pdbcheck)
2. Entropy Change. The case of a one-dimensional normal distribution
3. Entropy Change. The case of multinormal distribution
4. Dissociation Constant: Kd, Mol/L
5. Enthalpy change: delta (P), J
6. The thermal dissociation
7. Potential energy of electrostatic interaction between all amino acid residues taken in pairs: Wp, J
8. Potential energy of the lower vibrational level: Wp1, J
9. Potential energy of the upper vibrational level: Wp2, J
10. Step-by-step verification of the PDB file structure (link: http://biomarker.co.il/pdbcheck)
Additional calculated parameters:
PLX-5568
PLX5568 selectively inhibits a Raf kinase and slowed the cyst growth in a rat ADPKD model, but the renal function did not improve because of increased renal fibrosis. A novel kinase inhibitor targeted for the treatment of at least two major indications with unmet medical needs: pain as well as polycystic kidney disease (PKD). PLX5568 has demonstrated robust preclinical efficacy in multiple pain models, including neuropathic pain as well as acute and inflammatory pain. In addition, PLX5568 has demonstrated compelling efficacy in multiple preclinical models of PKD.
Mutations in the P loop of BRAF are present at relatively high frequencies in lung adenocarcinoma: ∼13% of the BRAF-mutant cases harbor BRAFG469A and ∼22% exhibit BRAFG466V
[Preclinical efficacy of a RAF inhibitor that evades paradoxical MAPK pathway activation in protein kinase BRAF-mutant lung cancer]
PLX5568 was obtained from Plexxikon, Berkeley.
In a Scaffold-Based Drug Discovery™ platform developed by Plexxikon, PLX5568 has been identified as selectively targeting a unique binding site of the Raf protein, potentially highly selective for Raf kinases.
In a Scaffold-Based Drug Discovery™ platform developed by Plexxikon, PLX5568 has been identified as selectively targeting a unique binding site of the Raf protein, potentially highly selective for Raf kinases.
[The Raf kinase inhibitor PLX5568 slows cyst proliferation in rat polycystic kidney disease but promotes renal and hepatic fibrosis]
BERKELEY, Calif., September 3, 2008--(Business Wire)--Plexxikon Inc. today announced that it has initiated a Phase 1 human clinical trial for PLX5568, a novel kinase inhibitor targeted for the treatment of at least two major indications with unmet medical needs: pain as well as polycystic kidney disease (PKD). PLX5568 has demonstrated robust preclinical efficacy in multiple pain models, including neuropathic pain as well as acute and inflammatory pain. In addition, PLX5568 has demonstrated compelling efficacy in multiple preclinical models of PKD.
PLX-5568
The B-Raf assays were run in 100x10E-6 M ATP
PLX5568 is a very selective and potent inhibitor of Raf kinase, a critical mediator of PKD pathology. PLX5568 has demonstrated impressive efficacy in orthologous models of both genetic forms of PKD, resulting in decreased cyst size and improved kidney function. models of PKD.
____________IC50
B-Raf___250nM
B-Raf___250nM


PLX5568 attenuated cell
proliferation and cyst growth in vitro.
proliferation and cyst growth in vitro.
MDCK cells were seeded in collagen I gels for 3 days and then treated with 0, 10, 100 or 1000 nM of PLX5568 for 8 days. Morphology and sizes of ~1100–1200 cysts per condition of six individual experiments were obtained and summarized as means 6 SEM. *P < 0.05.
Cell proliferation of human ADPKD cyst cells was stimulated with 100 lM 8-Br-cAMP in the presence or absence of different concentrations of PLX5568 as specified.
[The Raf kinase inhibitor PLX5568 slows cyst proliferation in rat polycystic kidney disease but promotes renal and hepatic fibrosis]

BRAF
PLX-5568
G506
S507
G509
[PLX-5568-BRAF]

G506A
Alive:2
adenomas
and adenocarcinomas
Alive:2
adenomas
and adenocarcinomas
S507L
Alive:0/4
nevi and melanomas
Alive:0/4
nevi and melanomas
G509A
Alive:6/4
nevi and melanomas
neoplasms, nos
transitional cell papillomas and carcinomas
squamous cell neoplasms
cystic, musinous and serous neoplasms
paragangliomas and glomus tumors
soft tissue tumors and nos
Alive:6/4
nevi and melanomas
neoplasms, nos
transitional cell papillomas and carcinomas
squamous cell neoplasms
cystic, musinous and serous neoplasms
paragangliomas and glomus tumors
soft tissue tumors and nos
G509R
Alive:3/2
nevi and melanomas
gliomas
squamous cell neoplasms
Alive:3/2
nevi and melanomas
gliomas
squamous cell neoplasms
D634N
Alive:13/4
Plasma cell tumor
adenomas and adenocarcinomas
acinal cell neolasm
mature b-cell limphomas
squamous cell neoplasms
nevi and melanomas
neoplasms, nos
Alive:13/4
Plasma cell tumor
adenomas and adenocarcinomas
acinal cell neolasm
mature b-cell limphomas
squamous cell neoplasms
nevi and melanomas
neoplasms, nos
G506E
Alive:8/2
nevi and melanomas
plasma cell tumors
squamous cell neoplasms
adenomas and adenocarcinomas
Alive:8/2
nevi and melanomas
plasma cell tumors
squamous cell neoplasms
adenomas and adenocarcinomas
G506V
Alive:3/4
Plasma cell tumor
adenomas and adenocarcinomas
cystic, musinous and serous neoplasms
Alive:3/4
Plasma cell tumor
adenomas and adenocarcinomas
cystic, musinous and serous neoplasms
G509V
Alive:5/1
adenomas and adenocarcinomas
Alive:5/1
adenomas and adenocarcinomas

G506R
Alive:0/1
plasma cell tumors
Alive:0/1
plasma cell tumors
S507L
Alive:0/4
nevi and melanomas
Alive:0/4
nevi and melanomas
D634G
Alive:5/3
plasma cell tumors
adenomas and adenocarcinomas
Alive:5/3
plasma cell tumors
adenomas and adenocarcinomas
G509E
Alive:2/0
nevi and melanomas
squamous cell neoplasms
Alive:2/0
nevi and melanomas
squamous cell neoplasms
N621S
Alive:4/3
adenomas and adenocarcinomas
cystic, musinous and serous neoplasms
nevi and melanomas
Alive:4/3
adenomas and adenocarcinomas
cystic, musinous and serous neoplasms
nevi and melanomas
The results of calculations of the effect of modification of the BRAF protein upon interaction with PLX-5568

The results of calculations of the effect of modification of the BRAF protein upon interaction with PLX-5568
Findings and Conclusions.
During preliminary calculations of the interaction of a small molecule PLX5568 with the BRAF protein, in which various replacements of amino acid residues taken from the cancer genomic atlas were performed, mutations in the BRAF protein were detected, which reduce the affinity of the interaction between the small chemical molecule and the BRAF protein (in red), and the matations in the BRAF protein that result in higher affinity are in green.
During preliminary calculations of the interaction of a small molecule PLX5568 with the BRAF protein, in which various replacements of amino acid residues taken from the cancer genomic atlas were performed, mutations in the BRAF protein were detected, which reduce the affinity of the interaction between the small chemical molecule and the BRAF protein (in red), and the matations in the BRAF protein that result in higher affinity are in green.
G506A
G509A
D634G
N621S
G509A
D634G
N621S
G506E/R
G506V
G507S
G509R
D634N
G506V
G507S
G509R
D634N
1. The fraction of non-dissociated molecules after the reaction and concentration protein-ligand complex
2. Entropy Change. The case of a one-dimensional normal distribution
3. Entropy Change. The case of multinormal distribution
4. Dissociation Constant: Kd, Mol/L
5. Enthalpy change: delta (P), J
6. The thermal dissociation
7. Potential energy of electrostatic interaction between all amino acid residues taken in pairs: Wp, J
8. Potential energy of the lower vibrational level: Wp1, J
9. Potential energy of the upper vibrational level: Wp2, J
10. Step-by-step verification of the PDB file structure (link: http://biomarker.co.il/pdbcheck)
2. Entropy Change. The case of a one-dimensional normal distribution
3. Entropy Change. The case of multinormal distribution
4. Dissociation Constant: Kd, Mol/L
5. Enthalpy change: delta (P), J
6. The thermal dissociation
7. Potential energy of electrostatic interaction between all amino acid residues taken in pairs: Wp, J
8. Potential energy of the lower vibrational level: Wp1, J
9. Potential energy of the upper vibrational level: Wp2, J
10. Step-by-step verification of the PDB file structure (link: http://biomarker.co.il/pdbcheck)
Additional calculated parameters:
PLX7904
C24H22F2N6O3S.
Next-generation Raf inhibitors, such as PLX7904 (PB04), effectively inhibit Raf signaling in BRaf(V600E) melanoma cells without paradoxical effects in wild-type cells. Furthermore, PLX7904 blocks the growth of vemurafenib-resistant BRaf(V600E) cells that express mutant NRAS. Acquired resistance to vemurafenib and dabrafenib is also frequently driven by expression of mutation BRaf splice variants
Next-generation Raf inhibitors, such as PLX7904 (PB04), effectively inhibit Raf signaling in BRaf(V600E) melanoma cells without paradoxical effects in wild-type cells. Furthermore, PLX7904 blocks the growth of vemurafenib-resistant BRaf(V600E) cells that express mutant NRAS. Acquired resistance to vemurafenib and dabrafenib is also frequently driven by expression of mutation BRaf splice variants

G509E
Alive:2/0
nevi and melanomas
squamous cell neoplasms
Alive:2/0
nevi and melanomas
squamous cell neoplasms
N621S
Alive:4/3
adenomas and adenocarcinomas
cystic, musinous and serous neoplasms
nevi and melanomas
Alive:4/3
adenomas and adenocarcinomas
cystic, musinous and serous neoplasms
nevi and melanomas
D634N
Alive:13/4
Plasma cell tumor
adenomas and adenocarcinomas
acinal cell neolasm
mature b-cell limphomas
squamous cell neoplasms
nevi and melanomas
neoplasms, nos
Alive:13/4
Plasma cell tumor
adenomas and adenocarcinomas
acinal cell neolasm
mature b-cell limphomas
squamous cell neoplasms
nevi and melanomas
neoplasms, nos
G509R
Alive:3/2
nevi and melanomas
gliomas
squamous cell
neoplasms
Alive:3/2
nevi and melanomas
gliomas
squamous cell
neoplasms
D634G
Alive:5/3
plasma cell tumors
adenomas and adenocarcinomas
Alive:5/3
plasma cell tumors
adenomas and adenocarcinomas
G509V
Alive:5/1
adenomas and adenocarcinomas
Alive:5/1
adenomas and adenocarcinomas
G509A
Alive:6/4
nevi and melanomas
neoplasms, nos
transitional cell papillomas and carcinomas
squamous cell neoplasms
cystic, musinous and serous neoplasms
paragangliomas and glomus tumors
soft tissue tumors and nos
Alive:6/4
nevi and melanomas
neoplasms, nos
transitional cell papillomas and carcinomas
squamous cell neoplasms
cystic, musinous and serous neoplasms
paragangliomas and glomus tumors
soft tissue tumors and nos
PLX7904 is a potent and selective BRAF inhibitor, with IC50 of appr 5 nM against BRAFV600E in mutant RAS exeported as “paradox breakers” that had no effect on MAPK pathway activation when suppressing mutant BRAF cells57 and PLX8394 also exhibited precise suppression of BRAF-V600 mutation without paradoxically enhancing the MAPK pathway in CRC.68 A
PLX7904 inhibits the in vitro growth of two melanoma cell lines (A375 and COLO829) and an additional human colorectal cancer cell line COLO205 that expresses BRAFV600E with IC50 values of 0.17 μM, 0.53 μM, and 0.16 μM, respectively, on a par with vemurafenib IC50 values in the same assays (0.33 μM, 0.69 μM, and 0.25 μM, respectively


PLX7904
The development of the next generation BRAFV600E inhibitors PLX7904 and its analogue PLX-8394, known as paradox breakers (PBs), the latter being in clinical trials in adult and paediatric patients with advanced BRAF-mutated tumours (NCT02428712), has opened new avenues in overcoming several mechanisms of resistance and controlling the paradoxical activation of the MAPK pathway
It is shown that PB PLX7904 and PLX8394 cause a more prolonged inhibition of the MAPK pathway and achieve a stronger proliferation blockage and reduced cell viability
It is shown that PB PLX7904 and PLX8394 cause a more prolonged inhibition of the MAPK pathway and achieve a stronger proliferation blockage and reduced cell viability

Cell survival for a panel of BRAFmt colon cancer cell lines treated with BRAF inhibitors paradox breakers (PBs) vs PLX4720. Effect on viability, 72 h post treatment with paradox breakers PLX7904 or PLX8394 vs. PLX4720 for the BRAFmut cell lines: RKO, HT29 and Colo-205, according to SRB assay A) IC50 values (μМ) for 72 h treatment with the three BRAFi
The evaluation of the new BRAF inhibitors-paradox breakers (PBs) PLX7904 was performed in cultures of colon cancer cell lines RKO, HT29 and Colo-205 that bear an heterozygous BRAFV600E mutation.
In BRAFV600E colorectal cancer, the BRAF mutant induces through the MAPK signalling the phosphorylation and subsequent the stabilization of the anti-apoptotic protein MCL-1 [26], and this mechanism of resistance to apoptosis can be suspended by simultaneous inhibition of MCL-1 and components of the MAPK pathway. This data indicate that simultaneously inhibition of MCL-1 and BRAFV600E might have synergistic effect in the treatment of BRAFV600E colorectal cancer
The results of calculations of the effect of modification of the BRAF protein upon interaction with PLX7904
[BRAF paradox breakers PLX8394, PLX7904 are more effective against BRAFV600Е CRC cells compared with the BRAF inhibitor PLX4720 and shown by detailed pathway analysis]

1. The fraction of non-dissociated molecules after the reaction and concentration protein-ligand complex
2. Entropy Change. The case of a one-dimensional normal distribution
3. Entropy Change. The case of multinormal distribution
4. Dissociation Constant: Kd, Mol/L
5. Enthalpy change: delta (P), J
6. The thermal dissociation
7. Potential energy of electrostatic interaction between all amino acid residues taken in pairs: Wp, J
8. Potential energy of the lower vibrational level: Wp1, J
9. Potential energy of the upper vibrational level: Wp2, J
10. Step-by-step verification of the PDB file structure (link: http://biomarker.co.il/pdbcheck)
2. Entropy Change. The case of a one-dimensional normal distribution
3. Entropy Change. The case of multinormal distribution
4. Dissociation Constant: Kd, Mol/L
5. Enthalpy change: delta (P), J
6. The thermal dissociation
7. Potential energy of electrostatic interaction between all amino acid residues taken in pairs: Wp, J
8. Potential energy of the lower vibrational level: Wp1, J
9. Potential energy of the upper vibrational level: Wp2, J
10. Step-by-step verification of the PDB file structure (link: http://biomarker.co.il/pdbcheck)
Findings and Conclusions.
During preliminary calculations of the interaction of a small molecule PLX7904 with the BRAF protein, in which various replacements of amino acid residues taken from the cancer genomic atlas were performed, mutations in the BRAF protein were detected, which reduce the affinity of the interaction between the small chemical molecule and the BRAF protein (in red), and the matations in the BRAF protein that result in higher affinity are in green.
During preliminary calculations of the interaction of a small molecule PLX7904 with the BRAF protein, in which various replacements of amino acid residues taken from the cancer genomic atlas were performed, mutations in the BRAF protein were detected, which reduce the affinity of the interaction between the small chemical molecule and the BRAF protein (in red), and the matations in the BRAF protein that result in higher affinity are in green.
Additional calculated parameters:
G509E/V600E
G509A/V600E
G509V/V600E
N621S/V600E
D634G/V600E
G509A/V600E
G509V/V600E
N621S/V600E
D634G/V600E
D634N/V600E
Double mutations
Double mutations
Sulfonamide
Sulfonamide-based B-Raf inhibitors have emerged as a class of effective agents in treating
patients with malignant melanoma harboring B-RafV600E, as exemplified by the approval of vemurafenib and dabrafenib.
patients with malignant melanoma harboring B-RafV600E, as exemplified by the approval of vemurafenib and dabrafenib.


Vemurafenib
2
2
1
Dabrafenib
3
3
R = Amino
6a
6a

Comparison of the activity and selectivity of compounds 1, 2, and 3.


Compound 1 was
equally potent toward both wild-type and mutant B-Raf in the biochemical assays where phosphorylation of MEK1 by the B-Raf proteins were measured
equally potent toward both wild-type and mutant B-Raf in the biochemical assays where phosphorylation of MEK1 by the B-Raf proteins were measured
Further investigations
revealed that, although compound 1 potently inhibited
down-stream ERK phosphorylation (p-ERK, IC50 = 2 nM) in A375 cells which harbor the B-RafV600E protein, it also activated the signaling pathway in the human pancreatic carcinoma MIA PaCa-2 cells which carry the B-RafWT protein (p-ERK, EC50 = 22 nM).
revealed that, although compound 1 potently inhibited
down-stream ERK phosphorylation (p-ERK, IC50 = 2 nM) in A375 cells which harbor the B-RafV600E protein, it also activated the signaling pathway in the human pancreatic carcinoma MIA PaCa-2 cells which carry the B-RafWT protein (p-ERK, EC50 = 22 nM).
Concurrent to our findings, similar results with other
second-generation B-Raf inhibitors appeared in the literature that also suggested that inhibition of B-RafWT may result in the seemingly paradoxical activation of the MAPK pathway by B-Raf inhibitors.9
One hypothesis that has been put forth is that, in the presence of high levels of activated Ras protein, the B-RafWT-inhibitor complex may induce hetero-dimer formation with C-Raf or other RAF family members.
[Purinylpyridinylamino-based DFG-in/aC-helix-out B-Raf inhibitors: Applying mutant versus wild-type B-Raf selectivity indices for compound profiling]
second-generation B-Raf inhibitors appeared in the literature that also suggested that inhibition of B-RafWT may result in the seemingly paradoxical activation of the MAPK pathway by B-Raf inhibitors.9
One hypothesis that has been put forth is that, in the presence of high levels of activated Ras protein, the B-RafWT-inhibitor complex may induce hetero-dimer formation with C-Raf or other RAF family members.
[Purinylpyridinylamino-based DFG-in/aC-helix-out B-Raf inhibitors: Applying mutant versus wild-type B-Raf selectivity indices for compound profiling]
The crystal structure confirmed
the expected mode of binding for the compound based on our initial design considerations, but it also presented an unexpected surprise in that the crystal lattice contained both an inhibitor-bound B-RafWT dimer and an inhibitor-bound B-RafWT monomer
the expected mode of binding for the compound based on our initial design considerations, but it also presented an unexpected surprise in that the crystal lattice contained both an inhibitor-bound B-RafWT dimer and an inhibitor-bound B-RafWT monomer


Biochemical scheme with interaction of wild-type BRAF proteins
The results of calculations of the effect of modification of the BRAF protein upon interaction with Sulfanomide


Findings and Conclusions.
During preliminary calculations of the interaction of a small molecule Sulfanomide with the BRAF protein, in which various replacements of amino acid residues taken from the cancer genomic atlas were performed, mutations in the BRAF protein were detected, which reduce the affinity of the interaction between the small chemical molecule and the BRAF protein (in red), and the matations in the BRAF protein that result in higher affinity are in green.
During preliminary calculations of the interaction of a small molecule Sulfanomide with the BRAF protein, in which various replacements of amino acid residues taken from the cancer genomic atlas were performed, mutations in the BRAF protein were detected, which reduce the affinity of the interaction between the small chemical molecule and the BRAF protein (in red), and the matations in the BRAF protein that result in higher affinity are in green.
V600G
G506E/R
G506V/L
G509A
D634G
G506E/R
G506V/L
G509A
D634G
V600E
V600M
K601E
G506A
N621S
V600M
K601E
G506A
N621S
1. The fraction of non-dissociated molecules after the reaction and concentration protein-ligand complex
2. Entropy Change. The case of a one-dimensional normal distribution
3. Entropy Change. The case of multinormal distribution
4. Dissociation Constant: Kd, Mol/L
5. Enthalpy change: delta (P), J
6. The thermal dissociation
7. Potential energy of electrostatic interaction between all amino acid residues taken in pairs: Wp, J
8. Potential energy of the lower vibrational level: Wp1, J
9. Potential energy of the upper vibrational level: Wp2, J
10. Step-by-step verification of the PDB file structure (link: http://biomarker.co.il/pdbcheck)
2. Entropy Change. The case of a one-dimensional normal distribution
3. Entropy Change. The case of multinormal distribution
4. Dissociation Constant: Kd, Mol/L
5. Enthalpy change: delta (P), J
6. The thermal dissociation
7. Potential energy of electrostatic interaction between all amino acid residues taken in pairs: Wp, J
8. Potential energy of the lower vibrational level: Wp1, J
9. Potential energy of the upper vibrational level: Wp2, J
10. Step-by-step verification of the PDB file structure (link: http://biomarker.co.il/pdbcheck)
Additional calculated parameters:
CNS292
Taken together, structural
evidence reveals extensive and specific interactions between CS292 and the ATP binding
pocket of the BRAF kinase domain, establishing CS292 as an ATP competitive inhibitor and
confirming its potent inhibitory properties against both BRAFWT and BRAFV600E.
evidence reveals extensive and specific interactions between CS292 and the ATP binding
pocket of the BRAF kinase domain, establishing CS292 as an ATP competitive inhibitor and
confirming its potent inhibitory properties against both BRAFWT and BRAFV600E.

CS292 Concentrarion ( M___ x 10)
-6
Agreement between calculated and experimental data




The most potent BRAF inhibitor, CS292, was also shown to have an 2-fold selectivity for BRAFV600E (IC50 = 0.21 мM) over the wild-type enzyme
CS292
Dose–response curves of CS292 from an ELISA-based BRAF kinase inhibition assay
CS292 binds to the ATP binding pocket of the kinase and is an ATP competitive inhibitor. The structure of the kinase–inhibitor complex also demonstrates that CS292 binds to BRAF in an active conformation and suggests a mechanism for regulation of BRAF by phosphorylation and BRAFV600E oncogene-induced activation.
[The Crystal Structure of BRAF in Complex with an Organoruthenium Inhibitor Reveals a Mechanism for Inhibition of an Active Form of BRAF Kinase]
[The Crystal Structure of BRAF in Complex with an Organoruthenium Inhibitor Reveals a Mechanism for Inhibition of an Active Form of BRAF Kinase]

CNS292
G506A/E/V/L
G509A
G509A
G506A/E///V640E
G509A//V640E
G509A//V640E
V640E
V640M
S507L
G509V
G509R
V640M
S507L
G509V
G509R
G506V/L//V640E
S507L//V640E
G509V//V640E
G509R//V640E
S507L//V640E
G509V//V640E
G509R//V640E
G506A/E//V640M
G509A//V640M
G509A//V640M
G506V/L//V640M
S507L//V640M
G509V//V640M
G509R//V640M
S507L//V640M
G509V//V640M
G509R//V640M

1. The fraction of non-dissociated molecules after the reaction and concentration protein-ligand complex
2. Entropy Change. The case of a one-dimensional normal distribution
3. Entropy Change. The case of multinormal distribution
4. Dissociation Constant: Kd, Mol/L
5. Enthalpy change: delta (P), J
6. The thermal dissociation
7. Potential energy of electrostatic interaction between all amino acid residues taken in pairs: Wp, J
8. Potential energy of the lower vibrational level: Wp1, J
9. Potential energy of the upper vibrational level: Wp2, J
10. Step-by-step verification of the PDB file structure (link: http://biomarker.co.il/pdbcheck)
2. Entropy Change. The case of a one-dimensional normal distribution
3. Entropy Change. The case of multinormal distribution
4. Dissociation Constant: Kd, Mol/L
5. Enthalpy change: delta (P), J
6. The thermal dissociation
7. Potential energy of electrostatic interaction between all amino acid residues taken in pairs: Wp, J
8. Potential energy of the lower vibrational level: Wp1, J
9. Potential energy of the upper vibrational level: Wp2, J
10. Step-by-step verification of the PDB file structure (link: http://biomarker.co.il/pdbcheck)
Findings and Conclusions.
During preliminary calculations of the interaction of a small molecule CS292 with the BRAF protein, in which various replacements of amino acid residues taken from the cancer genomic atlas were performed, mutations in the BRAF protein were detected, which reduce the affinity of the interaction between the small chemical molecule and the BRAF protein (in red), and the matations in the BRAF protein that result in higher affinity are in green.
During preliminary calculations of the interaction of a small molecule CS292 with the BRAF protein, in which various replacements of amino acid residues taken from the cancer genomic atlas were performed, mutations in the BRAF protein were detected, which reduce the affinity of the interaction between the small chemical molecule and the BRAF protein (in red), and the matations in the BRAF protein that result in higher affinity are in green.
Additional calculated parameters:
Double mutations:
Double mutations:
G506A/E/V/L
G509A
G509A
G506A/E///V640E
G509A//V640E
G509A//V640E
V640E
V640M
S507L
G509V
G509R
V640M
S507L
G509V
G509R
G506V/L//V640E
S507L//V640E
G509V//V640E
G509R//V640E
S507L//V640E
G509V//V640E
G509R//V640E
G506A/E//V640M
G509A//V640M
G509A//V640M
G506V/L//V640M
S507L//V640M
G509V//V640M
G509R//V640M
S507L//V640M
G509V//V640M
G509R//V640M
Single mutations:
Ponatinib
an FDA-approved drug, is an effective inhibitor of BRAF monomers and dimers. Ponatinib binds the BRAF dimer and stabilizes a distinct αC-helix conformation through interaction with a previously unrevealed allosteric site

Ponatinib
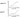

Agreement between calculated and experimental data



PHI1

IC50=10nM
IC50=323nM
IC50=323nM
In vitro kinase inhibition activity of Ponatinib.
Inhibition of kinase activity of BRAFV600E, BRAFWT and other tyrosine kinases targets by
Ponatinib using SelectScreen (Invitrogen) assay in the presence of 100 μM ATP. Half-maximal
inhibition values (IC50) in kinase activity by Ponatinib are measured. Data are mean ± SD of
two technical replicates from n=2 independent experiments.
Inhibition of kinase activity of BRAFV600E, BRAFWT and other tyrosine kinases targets by
Ponatinib using SelectScreen (Invitrogen) assay in the presence of 100 μM ATP. Half-maximal
inhibition values (IC50) in kinase activity by Ponatinib are measured. Data are mean ± SD of
two technical replicates from n=2 independent experiments.

In vitro kinase inhibition activity of PHI1.
Inhibition of kinase activity of BRAF and selected RTKs by PHI1 using SelectScreen (Invitrogen) assay in the presence of 100 μM ATP. Half-maximal inhibition (IC50) of kinase activities by PHI1 and Ponatinib (PON) are tabulated

In vitro BRAF kinase inhibition activity of RAF inhibitors.
BRAFWT and BRAFV600E kinase inhibition by selected αC-IN inhibitors (LY3009120, AZ-628, TAK-632) and αC-OUT inhibitors (Vemurafenib, Dabrafenib, Encorafenib), in comparison to BRAF inhibition by Ponatinib as reported here. Kinase activity was measured using SelectScreen (Invitrogen) assay in the presence of 100 μM ATP. Half-maximal inhibition values (IC50) in kinase activity are summarized on the table (bottom).
BRAFWT and BRAFV600E kinase inhibition by selected αC-IN inhibitors (LY3009120, AZ-628, TAK-632) and αC-OUT inhibitors (Vemurafenib, Dabrafenib, Encorafenib), in comparison to BRAF inhibition by Ponatinib as reported here. Kinase activity was measured using SelectScreen (Invitrogen) assay in the presence of 100 μM ATP. Half-maximal inhibition values (IC50) in kinase activity are summarized on the table (bottom).
Ponatinib is a RAF inhibitor
Kinase activity inhibition profiles of BRAFV600E and BRAFWT upon Ponatinib titration using SelectScreen assay [Inhibitors of BRAF dimers using an allosteric site]


Graph of a qualitative change in affinity during the interaction of Ponatinib with BRAF1(V600E) + additional mutations in the protein
Graph of a qualitative change in affinity during the interaction of Ponatinib with BRAF1(V600M) + additional mutations in the protein
Graph of a qualitative change in affinity during the interaction of Ponatinib with BRAF1 + additional mutations in the protein
nevi and melanomas
nevi and melanomas/ neoplasms// nos/transitional cell// papillomas and carcinomas// squamous cell neoplasms// cystic, musinous and serous neoplasms/ and glomus tumors/soft tissue tumors and nos
nevi and melanomas/ neoplasms// nos/transitional cell// papillomas and carcinomas// squamous cell neoplasms// cystic, musinous and serous neoplasms/ paragangliomas and glomus tumors/soft tissue tumors and nos
nevi and melanomas/ neoplasms// nos/transitional cell// papillomas and carcinomas// squamous cell neoplasms// cystic, musinous and serous neoplasms/ paragangliomas and glomus tumors/soft tissue tumors and nos
adenomas and adenocarcinomas
adenomas and adenocarcinomas
adenomas and adenocarcinomas
nevi and melanomas
adenomas and adenocarcinomas

Ponatinib
PHI1


You can enlarge + the Figures or view the interactive version on your laptop.
Findings and Conclusions.
During preliminary calculations of the interaction of a small molecule Ponatinib with the BRAF protein, in which various replacements of amino acid residues taken from the cancer genomic atlas were performed, mutations in the BRAF protein were detected, which reduce the affinity of the interaction between the small chemical molecule and the BRAF protein (in red), and the matations in the BRAF protein that result in higher affinity are in green.
During preliminary calculations of the interaction of a small molecule Ponatinib with the BRAF protein, in which various replacements of amino acid residues taken from the cancer genomic atlas were performed, mutations in the BRAF protein were detected, which reduce the affinity of the interaction between the small chemical molecule and the BRAF protein (in red), and the matations in the BRAF protein that result in higher affinity are in green.
V600E
V600M
G506E/V
G509R
G509V
V600M
G506E/V
G509R
G509V
G506A
S507L
G509A
G509V
S507L
G509A
G509V
1. The fraction of non-dissociated molecules after the reaction and concentration protein-ligand complex
2. Entropy Change. The case of a one-dimensional normal distribution
3. Entropy Change. The case of multinormal distribution
4. Dissociation Constant: Kd, Mol/L
5. Enthalpy change: delta (P), J
6. The thermal dissociation
7. Potential energy of electrostatic interaction between all amino acid residues taken in pairs: Wp, J
8. Potential energy of the lower vibrational level: Wp1, J
9. Potential energy of the upper vibrational level: Wp2, J
10. Step-by-step verification of the PDB file structure (link: http://biomarker.co.il/pdbcheck)
2. Entropy Change. The case of a one-dimensional normal distribution
3. Entropy Change. The case of multinormal distribution
4. Dissociation Constant: Kd, Mol/L
5. Enthalpy change: delta (P), J
6. The thermal dissociation
7. Potential energy of electrostatic interaction between all amino acid residues taken in pairs: Wp, J
8. Potential energy of the lower vibrational level: Wp1, J
9. Potential energy of the upper vibrational level: Wp2, J
10. Step-by-step verification of the PDB file structure (link: http://biomarker.co.il/pdbcheck)
Additional calculated parameters:
G506E/V//V600E
G509R//V600E
G509V//V600E
G509R//V600E
G509V//V600E
G506E/V//V600M
G509R//V600M
G509V//V600M
G509R//V600M
G509V//V600M
G506A//V600E
S507L//V600E
G509A//V600E
S507L//V600E
G509A//V600E
G506A//V600M
S507L//V600M
G509A//V600M
S507L//V600M
G509A//V600M
Double mutations:
Double mutations:
Findings and Conclusions.
During preliminary calculations of the interaction of a small molecule PHI1 with the BRAF protein, in which various replacements of amino acid residues taken from the cancer genomic atlas were performed, mutations in the BRAF protein were detected, which reduce the affinity of the interaction between the small chemical molecule and the BRAF protein (in red), and the matations in the BRAF protein that result in higher affinity are in green.
During preliminary calculations of the interaction of a small molecule PHI1 with the BRAF protein, in which various replacements of amino acid residues taken from the cancer genomic atlas were performed, mutations in the BRAF protein were detected, which reduce the affinity of the interaction between the small chemical molecule and the BRAF protein (in red), and the matations in the BRAF protein that result in higher affinity are in green.
V600E
V600M
G506E/V/L
G507L
G509R/V
V600M
G506E/V/L
G507L
G509R/V
G506A
G509A
G509A
1. The fraction of non-dissociated molecules after the reaction and concentration protein-ligand complex
2. Entropy Change. The case of a one-dimensional normal distribution
3. Entropy Change. The case of multinormal distribution
4. Dissociation Constant: Kd, Mol/L
5. Enthalpy change: delta (P), J
6. The thermal dissociation
7. Potential energy of electrostatic interaction between all amino acid residues taken in pairs: Wp, J
8. Potential energy of the lower vibrational level: Wp1, J
9. Potential energy of the upper vibrational level: Wp2, J
10. Step-by-step verification of the PDB file structure (link: http://biomarker.co.il/pdbcheck)
2. Entropy Change. The case of a one-dimensional normal distribution
3. Entropy Change. The case of multinormal distribution
4. Dissociation Constant: Kd, Mol/L
5. Enthalpy change: delta (P), J
6. The thermal dissociation
7. Potential energy of electrostatic interaction between all amino acid residues taken in pairs: Wp, J
8. Potential energy of the lower vibrational level: Wp1, J
9. Potential energy of the upper vibrational level: Wp2, J
10. Step-by-step verification of the PDB file structure (link: http://biomarker.co.il/pdbcheck)
Additional calculated parameters:
G506E/V/L//V600E
G507L//V600E
G509R//V600E
G509V//V600E
G507L//V600E
G509R//V600E
G509V//V600E
G506E/V/L//V600M
G507L//V600M
G509R//V600M
G509V//V600M
G507L//V600M
G509R//V600M
G509V//V600M
G506A//V600E
G509A//V600E
G509A//V600E
G506A//V600M
G509A//V600M
G509A//V600M
Double mutations:
Double mutations:
ATP prevents formation of RAFKD dimers
In the absence of nucleotide, two BRAFKD and two MEK1 molecules interact to form a hetero-tetrameric complex in solution mediated by the BRAFKD dimer. Inclusion of 100 μM ACP in solution disrupted BRAFKD dimers complexed with MEK1, resulting in a BRAFKD– MEK1 heterodimer complex, a direct consequence of ACP binding to BRAFKD consistent with a general negative regulatory role for ATP in blocking RAF dimer formation.

Any molecular event that destabilizes RAFKD dimers could be a negative regulatory mechanism. Physiological concentrations of ATP are known to disfavor formation of homodimers and heterodimers of RAFs. This presents a conundrum, whereby both ATP binding and RAFKD dimerization are necessary for activity, yet appear to be mutually antagonistic.
ACP binding induced an inactive conformation for the BRAFKD. This allosterically induced conformational change positions the N-lobe of one protomer away from the C-lobe of the other, and vice versa. Since the BRAFKD forms an N-to-C anti-parallel dimer, this conformational change rotates the RAFKD dimer interface away from both sides. This mechanism provides a structural rationale for the ability of ATP analogs to block RAFKD dimerization, as the residues that directly interact with ACP in BRAFKD are conserved between RAF isoforms.
ACP binding induced an inactive conformation for the BRAFKD. This allosterically induced conformational change positions the N-lobe of one protomer away from the C-lobe of the other, and vice versa. Since the BRAFKD forms an N-to-C anti-parallel dimer, this conformational change rotates the RAFKD dimer interface away from both sides. This mechanism provides a structural rationale for the ability of ATP analogs to block RAFKD dimerization, as the residues that directly interact with ACP in BRAFKD are conserved between RAF isoforms.
Addition of ACP did not disrupt the RAFKD(FCT)–MEK1–14-3-3 complexes, showing only slight shifts in the elution volume that were expected due to conformational
rearrangements. Thus, while ACP disrupted RAF–MEK tetramers, resulting in RAF–MEK heterodimers
rearrangements. Thus, while ACP disrupted RAF–MEK tetramers, resulting in RAF–MEK heterodimers
Oncogenic BRAF mutations promote dimerization. Since ATP binding negatively regulates RAF dimerization, and oncogenic mutations in BRAF activate the RAS–RAF–MEK–ERK pathway,
we speculated that the two opposing mechanisms may be connected.
we speculated that the two opposing mechanisms may be connected.
the top 12 most frequently observed oncogenic BRAF mutations
that account for >50% of all cancer associated BRAF mutations onto our ACP-induced, inactive monomeric RAF structure. All classes of mutations activate the pathway. Oncogenic mutations in BRAF act through a general mechanism of decoupling ATP binding from dimer destabilization, thus bypassing ATP-dependent negative regulation of BRAF kinase activity. Consistent with this hypothesis, ACP is unable to break BRAF dimers containing the V600E mutation
class I mutations V600
which increase kinase activity and act independently of dimerization
class II mutations
that increase kinase activity
class III mutations
that decrease kinase activity or render the kinase inactive and are dependent on dimerization
